Many people have heard of the modern wave of antibiotic resistant bacterial infections but a more pernicious form of drug resistance also manifests throughout the treatment of cancer. Therapies for cancer can largely be split into 2 separate categories; general cytotoxic and targeted treatments. Cytotoxic treatments are spatially targeted to the site of the cancer but are lethal to almost all cells connected by them whereas targeted therapies refer to the targeting of specific genes which are related to oncogenic pathways within the cell. In this particular study, we attempted to discover the dynamics of drug resistance to targeted combination therapy with BRAF inhibitors (BARFi) and MEK inhibitors (MEKi) in melanoma.
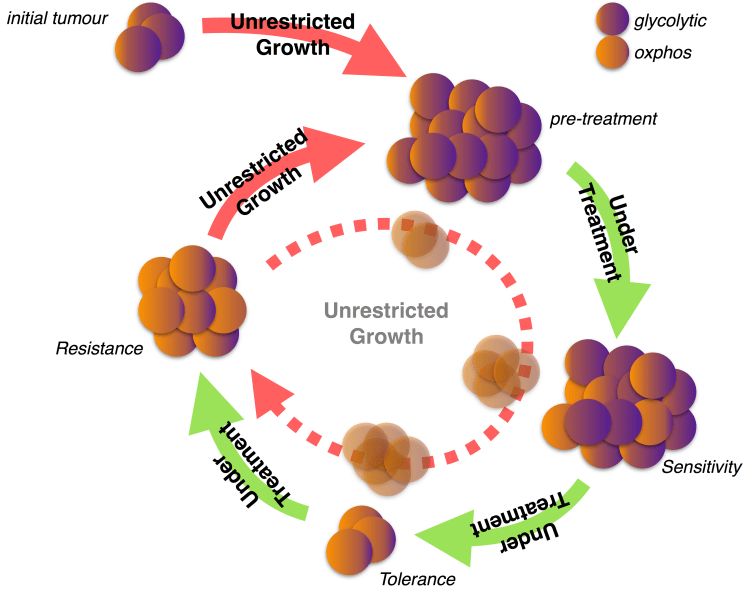
In order to understand why these treatments are used, one must firstly understand that cells may metabolise glucose through two pathways: Firstly, glucose is processed, through a series of enzymatic interactions, into a substance known as pyruvate. Pyruvate may then either be (a) converted in to acetyl coenzyme A (Acetyl-CoA) to feed the Krebs cycle and produce energy for the cell or (b) converted to lactate in a process yielding many byproducts which may be used to synthesise the lipid building blocks of the cell. In this study, we name pathway (a) the oxidative phosphorylation (oxphos) pathway, since this is the final step in the processing of pyruvate to energy, and pathway (b) glycolysis, since in the presence of oxygen this process is biologically termed aerobic glycolysis. Otto Warburg noticed that cancer cells were, to a greater extent than healthy cells, utilising glycolysis pathways in a phenomenon now known as the Warburg effect.
Therefore, in order to specifically target cancer cells, biochemists synthesised BRAFi and MEKi drugs to target BRAF and MEK, who are both implicated in the synthesis of enzymes used for glycolysis. In this study, we build a system of partial differential equations (PDEs) to understand the dynamics by which a bulk tumour may resist this targeted treatment and, as revealed by recent experiments, become re-sensitised to such therapies. Previous models of drug resistance have considered two populations of cells which are either resistant or sensitive to treatment, respectively. In order to understand metabolic dynamics, we have redescribed the biological tumour as existing in a distribution of states which are more or less glycolytic, and more or less oxphos. This new description allows us to explicate the continuous evolution of cells, on a population level, and permit random (diffusive) metabolic dynamics with a preferred (advective) centre of metabolism leaning towards glycolysis; as implied by the Warburg effect.
Our in silico model was able to predict characteristic death, tolerance, and regrowth tumour volume curves in response to multiple waves of therapy. We found that this was due to cancer cells, who diversified their metabolic activity in response to drug induced stress, going through a process of natural selection with BRAFi and MEKi degrading glycolytic subpopulations and allowing for the continued proliferation of the well adapted oxphos subpopulations. Moreover, we found that the response to a second wave of treatment with BRAFi and MEKi was more rapid than the first due to the primary wave of resistance leaving a residual population of oxphos cells, who were capable of proliferating in the presence of even large concentrations of the drug. This also appears to be amplified by the fact that both nutrients and drugs are entering the system by the same means — the local vasculature — and creating regions of high nutritional content co-localised with a strong selective pressure. Although these predictions match those made within recently obtained in vivo data, the justification for the prediction remains to be biologically verified.
(This manuscript can now be found on BioRxiv: https://www.biorxiv.org/content/early/2018/11/07/463877. Please make contact for any additional information.)
